Differential Exon Usage Analysis: DEXSeq
Last updated: 2021-09-25
Checks: 7 0
Knit directory: amnio-cell-free-RNA/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200224) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version e20c62d. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: .bpipe/
Ignored: analysis/obsolete_analysis/
Ignored: analysis/salmon-ruvseq-edger.nb.html
Ignored: code/.bpipe/
Ignored: code/.rnaseq-test.groovy.swp
Ignored: code/obsolete_analysis/
Ignored: data/.bpipe/
Ignored: data/190717_A00692_0021_AHLLHFDSXX/
Ignored: data/190729_A00692_0022_AHLLHFDSXX/
Ignored: data/190802_A00692_0023_AHLLHFDSXX/
Ignored: data/200612_A00692_0107_AHN3YCDMXX.tar
Ignored: data/200612_A00692_0107_AHN3YCDMXX/
Ignored: data/200626_A00692_0111_AHNJH7DMXX.tar
Ignored: data/200626_A00692_0111_AHNJH7DMXX/
Ignored: data/CMV-AF-database-corrected-oct-2020.csv
Ignored: data/CMV-AF-database-final-included-samples.csv
Ignored: data/GONE4.10.13.txt
Ignored: data/HK_exons.csv
Ignored: data/HK_exons.txt
Ignored: data/HK_genes.txt
Ignored: data/IPA molecule summary.xls
Ignored: data/IPA-molecule-summary.csv
Ignored: data/brain-development-geneset.txt
Ignored: data/deduped_rRNA_coverage.txt
Ignored: data/gene-transcriptome-analysis/
Ignored: data/hg38_rRNA.bed
Ignored: data/hg38_rRNA.saf
Ignored: data/ignore-overlap-mapping/
Ignored: data/ignore/
Ignored: data/joindata.csv
Ignored: data/metadata.csv
Ignored: data/rds/
Ignored: data/salmon-pilot-analysis/
Ignored: data/samples.csv
Ignored: data/star-genome-analysis/
Ignored: output/c2Ens.RData
Ignored: output/c5Ens.RData
Ignored: output/hEns.RData
Ignored: output/keggEns.RData
Untracked files:
Untracked: analysis/STAR-DEXSeq-exclude-US-ab.Rmd
Untracked: code/output.R
Untracked: code/rnaseq.salmon-quant.groovy
Untracked: code/rnaseq.star.count-exons.groovy
Untracked: code/rnaseq.star.groovy
Untracked: output/salmon-limma-voom-GO-exclude-CMV11.csv
Untracked: output/salmon-limma-voom-c2Cam-exclude-CMV11.csv
Untracked: output/salmon-limma-voom-c5Cam-exclude-CMV11.csv
Untracked: output/salmon-limma-voom-c5Cam.csv
Untracked: output/salmon-limma-voom-exclude-CMV11.csv
Untracked: output/salmon-limma-voom-hCam-exclude-CMV11.csv
Untracked: output/salmon-limma-voom-keggCam-exclude-CMV11.csv
Untracked: output/salmon-limma-voom.csv
Untracked: output/salmon-ruvseq-edger.csv
Untracked: output/star-fc-ruv-all-gsea-c2.csv
Untracked: output/star-fc-ruv-all-gsea-c5.csv
Untracked: output/star-fc-ruv-all.csv
Untracked: output/star-fc-ruv-no_us_ab-gsea-c2.csv
Untracked: output/star-fc-ruv-no_us_ab-gsea-c5.csv
Untracked: output/star-fc-ruv-no_us_ab.csv
Untracked: renv.lock
Unstaged changes:
Modified: .gitignore
Deleted: analysis/STAR-DEXSeq-Summed.Rmd
Deleted: analysis/salmon-BANDITS.Rmd
Deleted: analysis/salmon-DRIMseq.Rmd
Deleted: analysis/salmon-RUV-all.Rmd
Deleted: analysis/salmon-SatuRn.Rmd
Deleted: code/rnaseq-with-salmon-quant.groovy
Deleted: code/rnaseq.groovy
Deleted: code/salmon-quant-trimmed.groovy
Deleted: code/unmapped-pipe.groovy
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/STAR-DEXSeq.Rmd) and HTML (docs/STAR-DEXSeq.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | e20c62d | Jovana Maksimovic | 2021-09-25 | wflow_publish(c(“analysis/index.Rmd”, “analysis/salmon-limma-voom.Rmd”, |
| html | b85b1d7 | Jovana Maksimovic | 2021-09-17 | Build site. |
| Rmd | 153ebef | Jovana Maksimovic | 2021-09-17 | wflow_publish(c(“analysis/index.Rmd”, “analysis/STAR-DEXSeq.Rmd”)) |
Data import
readHTSeq <- function(files){
names(files) <- strsplit2(files, "_")[,6]
files <- files[grepl("CMV", names(files))]
tmp <- lapply(files, function(file){
read.delim(file, sep = "\t", row.names = 1, header = FALSE)
})
counts <- bind_cols(tmp)
colnames(counts) <- names(tmp)
rownames(counts) <- sub(":", ":E", rownames(counts))
counts
}
pe <- readHTSeq(list.files(here("data/star-genome-analysis/count-exons-pe"),
pattern="PE.txt$", full.names = TRUE))New names:
* V2 -> V2...1
* V2 -> V2...2
* V2 -> V2...3
* V2 -> V2...4
* V2 -> V2...5
* ...se <- readHTSeq(list.files(here("data/star-genome-analysis/count-exons-se"),
pattern="SE.txt$", full.names = TRUE))New names:
* V2 -> V2...1
* V2 -> V2...2
* V2 -> V2...3
* V2 -> V2...4
* V2 -> V2...5
* ...counts <- pe + se
counts <- counts[grepl("^ENSG", rownames(counts)),]
dim(counts)[1] 662662 28Load sample information.
| id | CMV_status | pair | sex | GA_at_amnio | indication |
|---|---|---|---|---|---|
| CMV2 | neg | M1 | F | 20 | no_us_ab |
| CMV1 | pos | M1 | F | 21 | no_us_ab |
| CMV4 | pos | M2 | M | 21 | no_us_ab |
| CMV3 | neg | M2 | M | 22 | no_us_ab |
| CMV10 | neg | NC2 | F | 20 | us_ab |
| CMV11 | pos | NC1 | F | 19 | us_ab |
| CMV19 | pos | NC2 | F | 18 | no_us_ab |
| CMV35 | neg | L5 | M | 21 | no_us_ab |
| CMV30 | pos | L1 | F | 21 | no_us_ab |
| CMV31 | neg | L1 | F | 21 | no_us_ab |
| CMV8 | neg | L2 | F | 23 | no_us_ab |
| CMV9 | pos | L2 | F | 23 | no_us_ab |
| CMV26 | pos | L3 | F | 22 | no_us_ab |
| CMV56 | neg | L3 | F | 21 | no_us_ab |
| CMV14 | neg | L4 | F | 21 | no_us_ab |
| CMV15 | pos | L4 | F | 22 | no_us_ab |
| CMV20 | pos | L5 | M | 21 | no_us_ab |
| CMV51 | neg | L6 | M | 22 | no_us_ab |
| CMV57 | pos | L6 | M | 21 | no_us_ab |
| CMV58 | pos | L7 | M | 20 | no_us_ab |
| CMV60 | neg | L7 | M | 20 | no_us_ab |
| CMV52 | pos | L8 | M | 22 | no_us_ab |
| CMV61 | neg | L8 | M | 22 | no_us_ab |
| CMV54 | neg | L9 | F | 21 | no_us_ab |
| CMV53 | pos | L9 | F | 21 | us_ab |
| CMV21 | neg | NC1 | F | 21 | no_us_ab |
Only retain paired samples with clinical information for downstream analysis.
int <- intersect(colnames(counts), targets$id)
targets <- targets[match(int, targets$id),]
counts <- counts[,match(int, colnames(counts))]
head(counts) %>% knitr::kable()| CMV30 | CMV31 | CMV8 | CMV9 | CMV26 | CMV14 | CMV15 | CMV20 | CMV21 | CMV1 | CMV2 | CMV3 | CMV4 | CMV10 | CMV11 | CMV19 | CMV35 | CMV51 | CMV52 | CMV53 | CMV54 | CMV56 | CMV57 | CMV58 | CMV60 | CMV61 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ENSG00000000003.15:E001 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ENSG00000000003.15:E002 | 33 | 34 | 21 | 45 | 46 | 41 | 36 | 35 | 38 | 45 | 38 | 19 | 43 | 21 | 14 | 49 | 25 | 23 | 24 | 38 | 33 | 21 | 38 | 27 | 15 | 14 |
| ENSG00000000003.15:E003 | 7 | 13 | 3 | 6 | 9 | 11 | 14 | 6 | 6 | 13 | 12 | 5 | 9 | 6 | 4 | 10 | 5 | 3 | 4 | 5 | 7 | 3 | 9 | 4 | 7 | 2 |
| ENSG00000000003.15:E004 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ENSG00000000003.15:E005 | 6 | 10 | 3 | 7 | 6 | 6 | 10 | 3 | 6 | 3 | 10 | 5 | 7 | 3 | 1 | 6 | 5 | 4 | 4 | 3 | 8 | 3 | 4 | 1 | 4 | 1 |
| ENSG00000000003.15:E006 | 8 | 6 | 3 | 7 | 2 | 4 | 8 | 3 | 6 | 5 | 10 | 5 | 6 | 5 | 3 | 3 | 4 | 3 | 4 | 0 | 5 | 2 | 4 | 3 | 3 | 1 |
DEXSeq analysis
Create the sample table.
sampleTable <- data.frame(row.names = targets$id,
condition = targets$CMV_status,
pair = targets$pair)
head(sampleTable) %>% knitr::kable()| condition | pair | |
|---|---|---|
| CMV30 | pos | L1 |
| CMV31 | neg | L1 |
| CMV8 | neg | L2 |
| CMV9 | pos | L2 |
| CMV26 | pos | L3 |
| CMV14 | neg | L4 |
Setup the data. Compare exon usage between CMV negative and positive samples. Sample pairing is taken into account.
formulaFullModel = ~ sample + exon + pair:exon + condition:exon
formulaReducedModel = ~ sample + exon + pair:exon
out <- here("data/rds/DEXSeq.rds")
if(!file.exists(out)){
splitted <- strsplit(rownames(counts), ":")
exons <- sapply(splitted, "[[", 2)
genesrle <- sapply(splitted, "[[", 1)
flatGff <- list.files(here("data/star-genome-analysis"),
pattern="DEXSeq.chr.gff$", full.names = TRUE)
aggregates <- read.delim(flatGff, stringsAsFactors = FALSE,
header = FALSE)
colnames(aggregates) <- c("chr", "source", "class", "start",
"end", "ex", "strand", "ex2", "attr")
aggregates$strand <- gsub("\\.", "*", aggregates$strand)
aggregates <- aggregates[which(aggregates$class == "exonic_part"), ]
aggregates$attr <- gsub("\"|=|;", "", aggregates$attr)
aggregates$gene_id <- sub(".*gene_id\\s(\\S+).*", "\\1",
aggregates$attr)
transcripts <- gsub(".*transcripts\\s(\\S+).*", "\\1",
aggregates$attr)
transcripts <- strsplit(transcripts, "\\+")
exonids <- gsub(".*exonic_part_number\\s(\\S+).*", "\\1",
aggregates$attr)
exoninfo <- GRanges(as.character(aggregates$chr),
IRanges(start = aggregates$start,
end = aggregates$end),
strand = aggregates$strand)
names(exoninfo) <- paste(aggregates$gene_id, exonids,
sep = ":E")
names(transcripts) <- rownames(exoninfo)
matching <- match(rownames(counts), names(exoninfo))
dxd <- DEXSeqDataSet(
as.matrix(counts),
sampleData = sampleTable,
design = ~ sample + exon + condition:exon,
featureID = exons,
groupID = genesrle,
exoninfo[matching],
transcripts[matching])
} else {
dxd <- readRDS(file = out)
}if(!file.exists(out)){
dxd = estimateSizeFactors( dxd )
} Estimate disperions. Plot.
if(!file.exists(out)){
BPPARAM = MulticoreParam(min(26, multicoreWorkers()))
dxd = estimateDispersions( dxd,
BPPARAM=BPPARAM,
formula = formulaFullModel )
}
plotDispEsts( dxd )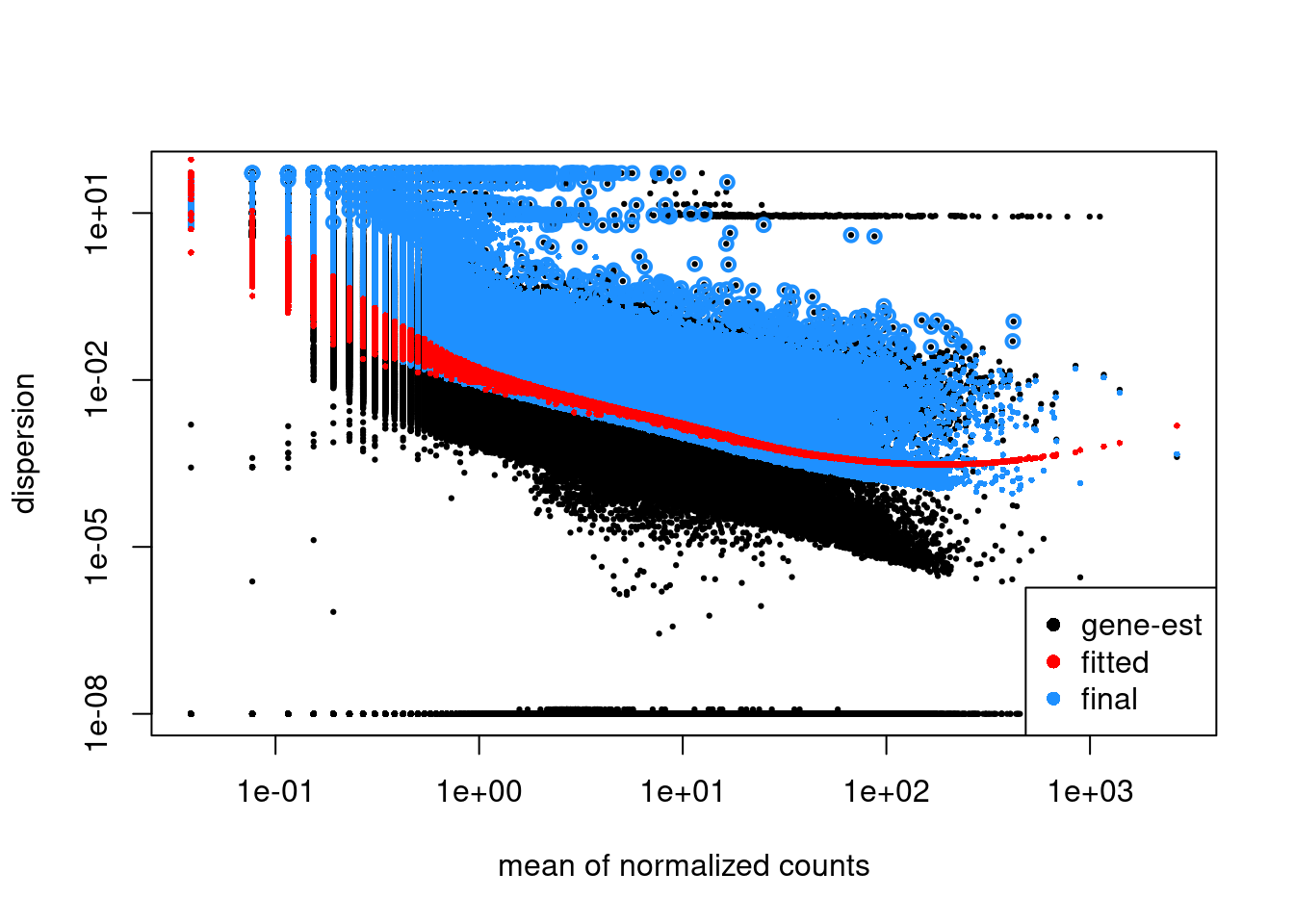
| Version | Author | Date |
|---|---|---|
| b85b1d7 | Jovana Maksimovic | 2021-09-17 |
Test for differential exon usage.
if(!file.exists(out)){
dxd = testForDEU( dxd,
BPPARAM=BPPARAM,
reducedModel = formulaReducedModel,
fullModel = formulaFullModel )
}Estimate exon fold changes.
if(!file.exists(out)){
dxd = estimateExonFoldChanges(dxd, BPPARAM=BPPARAM )
saveRDS(dxd, file = out)
} Number of statistically significant differentially used exons.
dxr1 = DEXSeqResults( dxd )
table ( dxr1$padj < 0.1 )
FALSE TRUE
282572 17 Number of genes with statistically significant differential exon usage.
table ( tapply( dxr1$padj < 0.1, dxr1$groupID, any ) )
FALSE TRUE
1034 13 MA plots of differential exons usage. Significant exons are shown in red.
DEXSeq::plotMA( dxr1, cex=0.8 )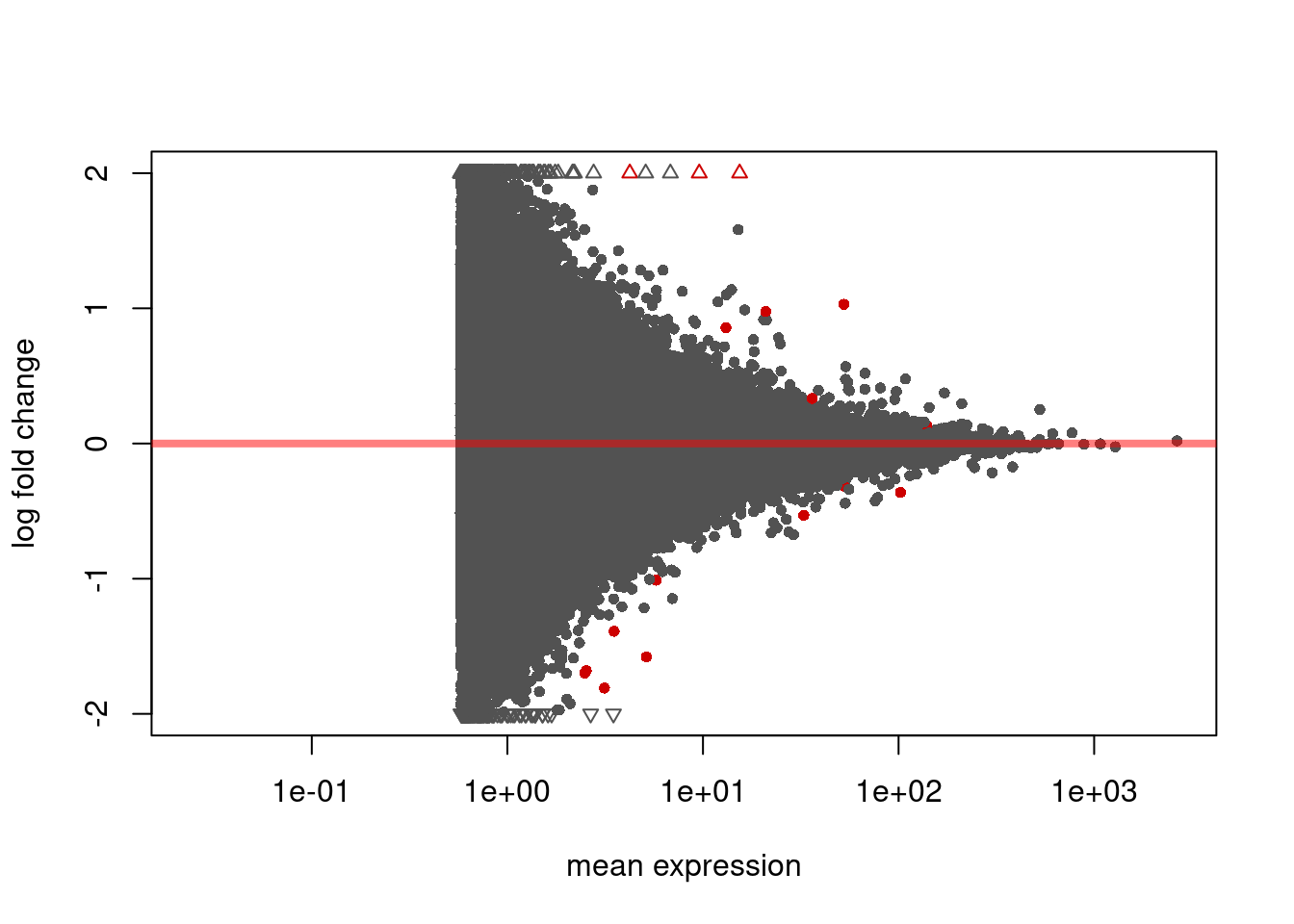
| Version | Author | Date |
|---|---|---|
| b85b1d7 | Jovana Maksimovic | 2021-09-17 |
Annotate results with gene symbols. Display most statistically significant differentially used exons.
deg <- read.csv(here("output/star-fc-ruv-all.csv"))
dxr1[which(dxr1$padj < 0.1),] %>% data.frame %>%
dplyr::arrange(groupID, padj) %>%
mutate(symbol = NA, .before = groupID) %>%
mutate(deg = NA, .after = symbol)-> topDex
for(i in 1:nrow(topDex)){
keys <- gsub("\\.[0-9]*", "",
strsplit(topDex$groupID[i], "+",
fixed=TRUE)[[1]])
symbol <- select(org.Hs.eg.db, keys = keys,
columns = c("ENTREZID", "SYMBOL"),
keytype = "ENSEMBL")$SYMBOL
topDex$symbol[i] <- paste(symbol, collapse = "+")
topDex$deg[i] <- any(keys %in% deg$Ensembl)
}'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columns
'select()' returned 1:1 mapping between keys and columnstopDex %>% knitr::kable()| symbol | deg | groupID | featureID | exonBaseMean | dispersion | stat | pvalue | padj | neg | pos | log2fold_pos_neg | genomicData.seqnames | genomicData.start | genomicData.end | genomicData.width | genomicData.strand | countData.CMV30 | countData.CMV31 | countData.CMV8 | countData.CMV9 | countData.CMV26 | countData.CMV14 | countData.CMV15 | countData.CMV20 | countData.CMV21 | countData.CMV1 | countData.CMV2 | countData.CMV3 | countData.CMV4 | countData.CMV10 | countData.CMV11 | countData.CMV19 | countData.CMV35 | countData.CMV51 | countData.CMV52 | countData.CMV53 | countData.CMV54 | countData.CMV56 | countData.CMV57 | countData.CMV58 | countData.CMV60 | countData.CMV61 | transcripts | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ENSG00000107959.16:E024 | PITRM1 | FALSE | ENSG00000107959.16 | E024 | 5.770499 | 0.0019295 | 20.50877 | 5.9e-06 | 0.0986710 | 0.9339937 | 0.6782048 | -1.0108556 | chr10 | 3148171 | 3148291 | 121 | - | 4 | 12 | 8 | 10 | 4 | 13 | 5 | 5 | 6 | 3 | 7 | 7 | 5 | 9 | 2 | 3 | 13 | 11 | 4 | 1 | 6 | 4 | 1 | 4 | 10 | 3 | ENST0000…. |
| ENSG00000136872.20:E020 | ALDOB | TRUE | ENSG00000136872.20 | E020 | 32.767507 | 0.0004712 | 21.38854 | 3.8e-06 | 0.0662329 | 1.5665267 | 1.4119173 | -0.5309091 | chr9 | 101430776 | 101430897 | 122 | - | 27 | 39 | 31 | 41 | 23 | 18 | 15 | 32 | 24 | 17 | 22 | 25 | 63 | 57 | 27 | 142 | 29 | 24 | 8 | 52 | 23 | 16 | 57 | 21 | 16 | 5 | ENST0000…. |
| ENSG00000141971.13+ENSG00000282851.2:E040 | MVB12A+BISPR | FALSE | ENSG00000141971.13+ENSG00000282851.2 | E040 | 4.225363 | 0.0028248 | 35.46654 | 0.0e+00 | 0.0002444 | 0.3123345 | 0.8104020 | 2.3754145 | chr19 | 17415200 | 17415656 | 457 | + | 2 | 1 | 1 | 25 | 1 | 2 | 7 | 4 | 0 | 3 | 0 | 1 | 7 | 2 | 6 | 17 | 0 | 1 | 0 | 9 | 2 | 0 | 7 | 5 | 1 | 1 | ENST0000…. |
| ENSG00000163913.12:E012 | IFT122 | FALSE | ENSG00000163913.12 | E012 | 3.512354 | 0.0027761 | 21.39059 | 3.7e-06 | 0.0662329 | 0.7845879 | 0.4688625 | -1.3888995 | chr3 | 129451973 | 129451998 | 26 | + | 2 | 7 | 7 | 1 | 3 | 7 | 6 | 0 | 5 | 3 | 7 | 6 | 3 | 8 | 0 | 0 | 5 | 9 | 1 | 0 | 1 | 9 | 4 | 2 | 3 | 3 | ENST0000…. |
| ENSG00000167461.12+ENSG00000196684.12:E019 | RAB8A+HSH2D | TRUE | ENSG00000167461.12+ENSG00000196684.12 | E019 | 52.596622 | 0.0005220 | 22.57256 | 2.0e-06 | 0.0476527 | 1.7631957 | 1.6701341 | -0.3152086 | chr19 | 16132320 | 16133010 | 691 | + | 42 | 58 | 41 | 69 | 55 | 53 | 47 | 57 | 53 | 65 | 73 | 50 | 65 | 67 | 28 | 85 | 64 | 59 | 36 | 72 | 60 | 63 | 57 | 55 | 26 | 24 | ENST0000…. |
| ENSG00000171793.16:E030 | CTPS1 | FALSE | ENSG00000171793.16 | E030 | 2.487082 | 0.0039669 | 23.14166 | 1.5e-06 | 0.0386619 | 0.7006737 | 0.3499062 | -1.6987830 | chr1 | 40997394 | 40997526 | 133 | + | 2 | 5 | 5 | 2 | 1 | 3 | 5 | 1 | 4 | 2 | 6 | 3 | 0 | 7 | 0 | 1 | 8 | 7 | 0 | 0 | 4 | 3 | 2 | 1 | 1 | 1 | ENST0000…. |
| ENSG00000197249.14:E016 | SERPINA1 | TRUE | ENSG00000197249.14 | E016 | 102.353104 | 0.0059674 | 23.67040 | 1.1e-06 | 0.0323076 | 2.0440776 | 1.9363302 | -0.3616374 | chr14 | 94383003 | 94383170 | 168 | - | 120 | 93 | 38 | 162 | 112 | 116 | 55 | 68 | 99 | 60 | 86 | 51 | 189 | 111 | 93 | 243 | 97 | 131 | 24 | 175 | 128 | 74 | 164 | 110 | 69 | 19 | ENST0000…. |
| ENSG00000204520.14+ENSG00000288587.1+ENSG00000206337.12:E040 | MICA+NA+HCP5 | TRUE | ENSG00000204520.14+ENSG00000288587.1+ENSG00000206337.12 | E040 | 15.394350 | 0.2341415 | 35.65253 | 0.0e+00 | 0.0002444 | 0.5340691 | 1.2916441 | 2.9398796 | chr6 | 31465889 | 31472408 | 6520 | + | 4 | 0 | 1 | 66 | 15 | 12 | 11 | 17 | 0 | 26 | 1 | 0 | 31 | 1 | 16 | 44 | 2 | 2 | 14 | 58 | 0 | 1 | 30 | 25 | 1 | 1 | ENST0000…. |
| ENSG00000204520.14+ENSG00000288587.1+ENSG00000206337.12:E021 | MICA+NA+HCP5 | TRUE | ENSG00000204520.14+ENSG00000288587.1+ENSG00000206337.12 | E021 | 3.135158 | 0.0545628 | 30.60043 | 0.0e+00 | 0.0017918 | 0.8275723 | 0.4205336 | -1.8088396 | chr6 | 31415012 | 31415259 | 248 | + | 3 | 3 | 3 | 0 | 6 | 2 | 1 | 7 | 2 | 3 | 7 | 5 | 5 | 4 | 3 | 2 | 7 | 8 | 1 | 1 | 3 | 6 | 1 | 2 | 0 | 2 | ENST0000…. |
| ENSG00000204520.14+ENSG00000288587.1+ENSG00000206337.12:E038 | MICA+NA+HCP5 | TRUE | ENSG00000204520.14+ENSG00000288587.1+ENSG00000206337.12 | E038 | 9.571713 | 0.2037536 | 25.86615 | 4.0e-07 | 0.0165797 | 0.4531283 | 1.0893134 | 2.6173800 | chr6 | 31464285 | 31465698 | 1414 | + | 4 | 2 | 0 | 33 | 4 | 9 | 11 | 7 | 3 | 13 | 1 | 0 | 14 | 0 | 12 | 39 | 0 | 1 | 9 | 30 | 1 | 0 | 25 | 16 | 0 | 0 | ENST0000…. |
| ENSG00000204520.14+ENSG00000288587.1+ENSG00000206337.12:E020 | MICA+NA+HCP5 | TRUE | ENSG00000204520.14+ENSG00000288587.1+ENSG00000206337.12 | E020 | 2.528471 | 0.0290812 | 25.21149 | 5.0e-07 | 0.0181476 | 0.7158093 | 0.3635786 | -1.6802196 | chr6 | 31412325 | 31412460 | 136 | + | 3 | 4 | 3 | 0 | 4 | 1 | 3 | 1 | 4 | 3 | 2 | 4 | 2 | 4 | 3 | 1 | 4 | 2 | 1 | 3 | 3 | 3 | 1 | 3 | 2 | 2 | ENST0000…. |
| ENSG00000204520.14+ENSG00000288587.1+ENSG00000206337.12:E019 | MICA+NA+HCP5 | TRUE | ENSG00000204520.14+ENSG00000288587.1+ENSG00000206337.12 | E019 | 5.138723 | 0.1432086 | 23.78031 | 1.1e-06 | 0.0323076 | 0.9908302 | 0.5958717 | -1.5785515 | chr6 | 31411947 | 31412225 | 279 | + | 2 | 6 | 14 | 2 | 10 | 6 | 6 | 5 | 7 | 7 | 6 | 5 | 10 | 7 | 4 | 2 | 6 | 9 | 2 | 4 | 6 | 3 | 5 | 4 | 5 | 0 | ENST0000…. |
| ENSG00000232110.8+ENSG00000119922.10:E013 | NA+IFIT2 | TRUE | ENSG00000232110.8+ENSG00000119922.10 | E013 | 36.126813 | 0.0005825 | 21.43066 | 3.7e-06 | 0.0662329 | 1.4912635 | 1.5885707 | 0.3328708 | chr10 | 89305962 | 89307372 | 1411 | + | 40 | 47 | 26 | 62 | 55 | 27 | 57 | 52 | 19 | 54 | 30 | 23 | 49 | 24 | 23 | 83 | 22 | 29 | 36 | 37 | 27 | 38 | 33 | 59 | 20 | 12 | ENST0000…. |
| ENSG00000234745.11+ENSG00000204525.16:E033 | HLA-B+HLA-C | TRUE | ENSG00000234745.11+ENSG00000204525.16 | E033 | 20.949796 | 0.0006547 | 37.93090 | 0.0e+00 | 0.0002071 | 1.0047755 | 1.2771857 | 0.9768858 | chr6 | 31353875 | 31354296 | 422 | - | 9 | 6 | 9 | 76 | 16 | 6 | 22 | 21 | 4 | 18 | 2 | 11 | 32 | 4 | 33 | 66 | 4 | 11 | 7 | 58 | 6 | 2 | 47 | 29 | 2 | 6 | ENST0000…. |
| ENSG00000242540.3+ENSG00000176887.7:E001 | NA+SOX11 | FALSE | ENSG00000242540.3+ENSG00000176887.7 | E001 | 139.526610 | 0.0002662 | 22.16199 | 2.5e-06 | 0.0544715 | 2.1383381 | 2.1770663 | 0.1295536 | chr2 | 5692384 | 5696219 | 3836 | + | 206 | 200 | 150 | 110 | 204 | 159 | 240 | 208 | 201 | 169 | 231 | 124 | 207 | 177 | 48 | 108 | 188 | 156 | 100 | 53 | 118 | 170 | 115 | 155 | 129 | 62 | ENST0000…. |
| ENSG00000254641.1+ENSG00000285338.1+ENSG00000166337.10+ENSG00000166340.17:E040 | NA+NA+TAF10+TPP1 | TRUE | ENSG00000254641.1+ENSG00000285338.1+ENSG00000166337.10+ENSG00000166340.17 | E040 | 52.561042 | 0.0272751 | 30.68357 | 0.0e+00 | 0.0017918 | 1.5341271 | 1.8375419 | 1.0295998 | chr11 | 6613304 | 6614600 | 1297 | - | 51 | 58 | 22 | 122 | 60 | 26 | 30 | 52 | 35 | 49 | 32 | 40 | 91 | 29 | 38 | 129 | 45 | 52 | 17 | 144 | 39 | 28 | 74 | 54 | 28 | 14 | ENST0000…. |
| ENSG00000259529.2+ENSG00000213928.9+ENSG00000092098.17:E087 | NA+IRF9+RNF31 | FALSE | ENSG00000259529.2+ENSG00000213928.9+ENSG00000092098.17 | E087 | 13.117604 | 0.0009589 | 25.64341 | 4.0e-07 | 0.0165797 | 0.9883130 | 1.2256805 | 0.8564411 | chr14 | 24162144 | 24162280 | 137 | + | 13 | 9 | 5 | 23 | 14 | 6 | 34 | 19 | 11 | 25 | 7 | 10 | 25 | 9 | 10 | 17 | 9 | 8 | 12 | 11 | 10 | 8 | 22 | 26 | 8 | 7 | ENST0000…. |
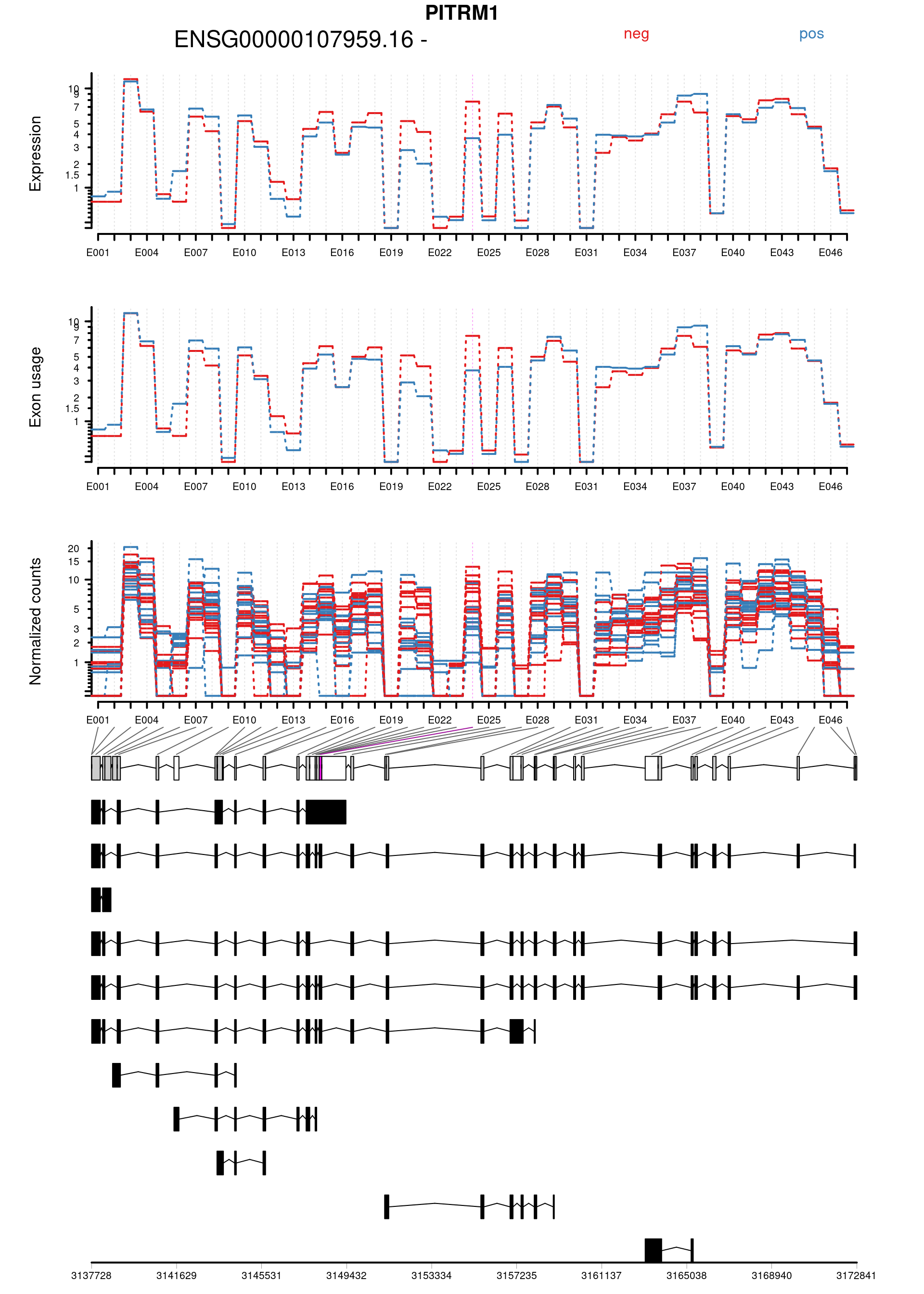
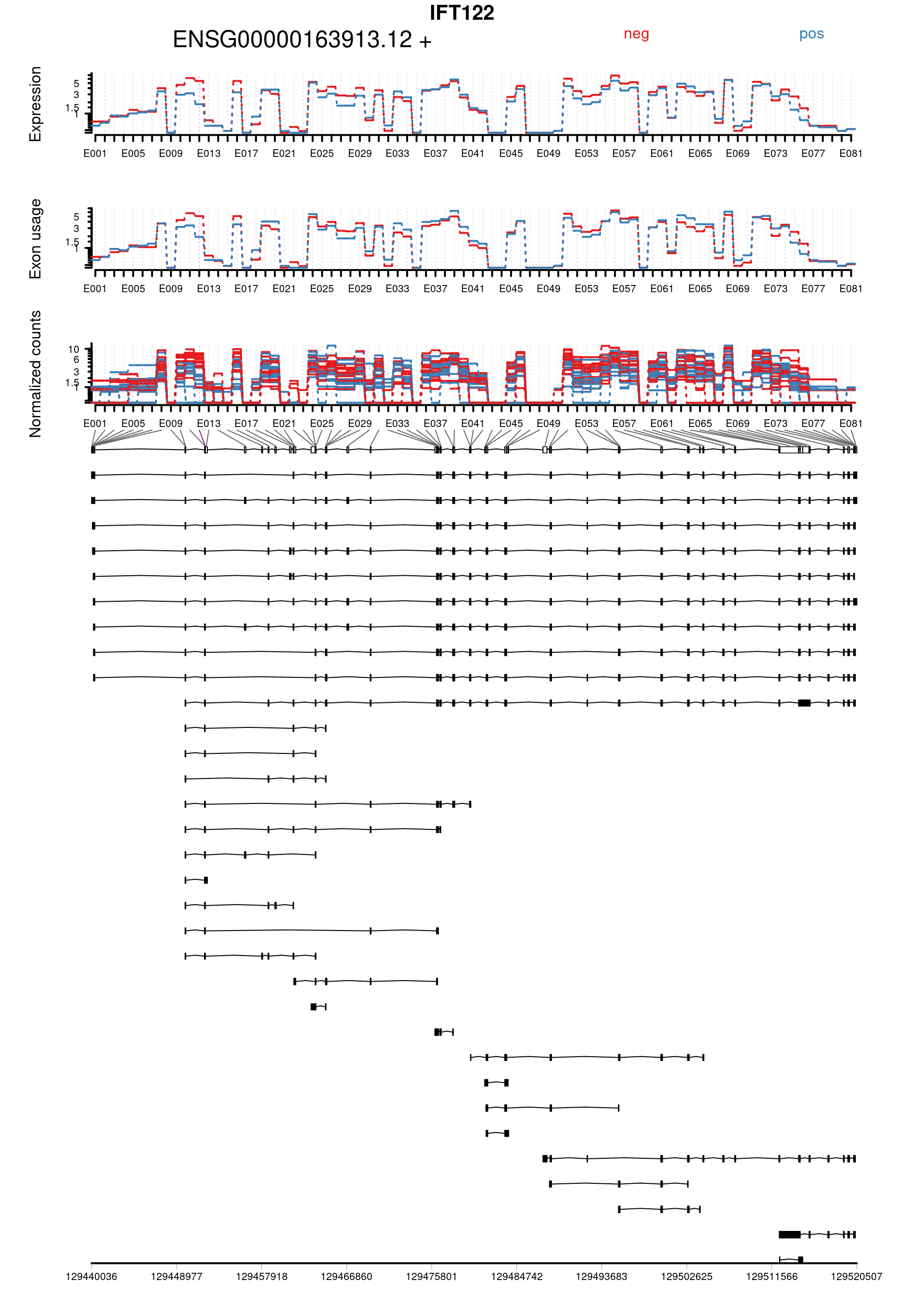
Plot genes with statistically significant differential exon usage. Exclude exons belonging to multiple genes.
keep <- !grepl("+",topDex$groupID, fixed = TRUE)
dexGenes <- unique(topDex$groupID[keep])
dexSymbols <- unique(topDex$symbol[keep])
par(oma = c(1,1,2,1))
for(i in 1:length(dexGenes)){
plotDEXSeq( dxr1, dexGenes[i],
legend=TRUE, cex.axis=1, cex=1, lwd=2,
displayTranscripts = TRUE, splicing = TRUE,
expression = TRUE, norCounts = TRUE)
title(main = dexSymbols[i], outer = TRUE, cex.main = 2)
}
| Version | Author | Date |
|---|---|---|
| b85b1d7 | Jovana Maksimovic | 2021-09-17 |

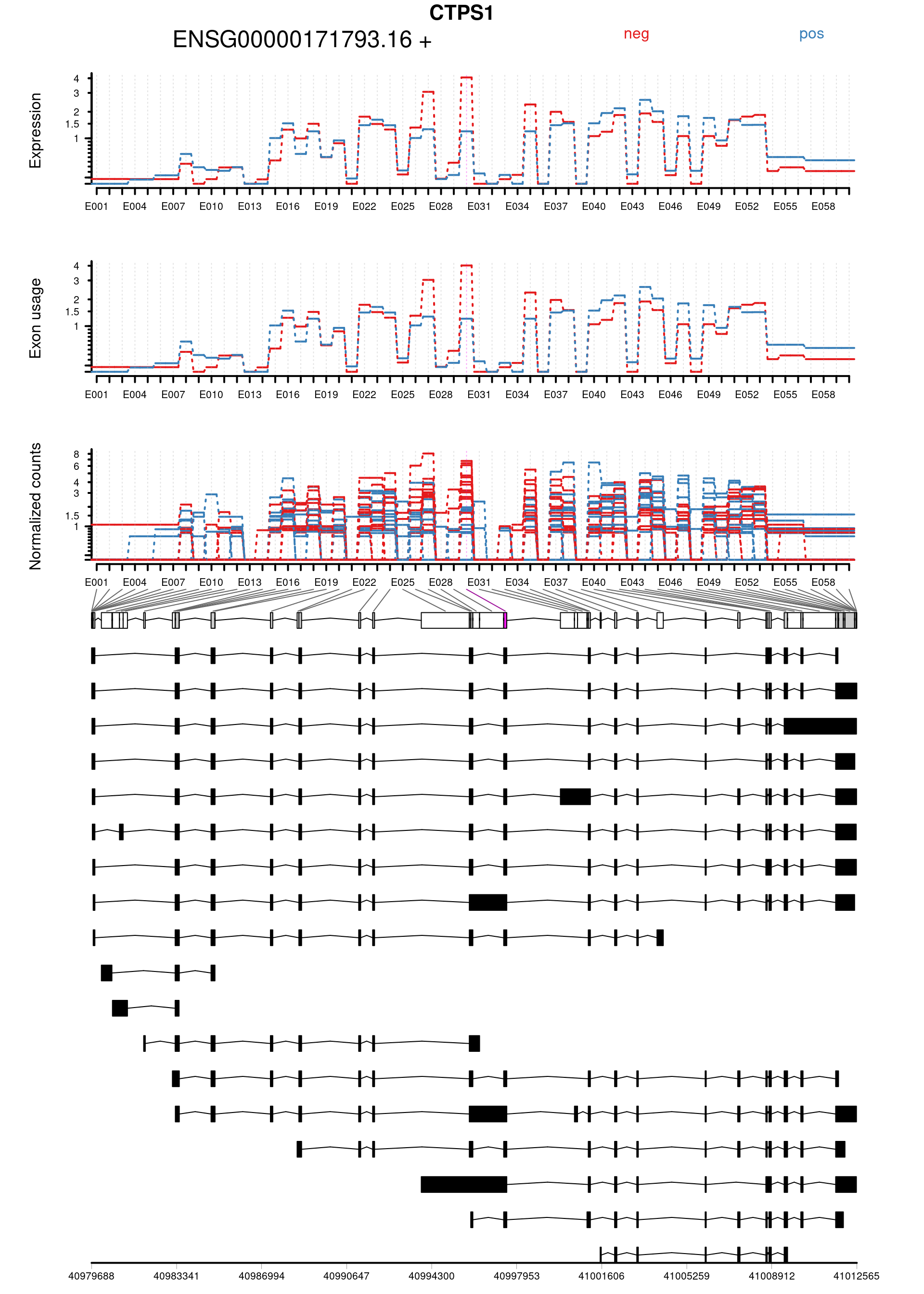
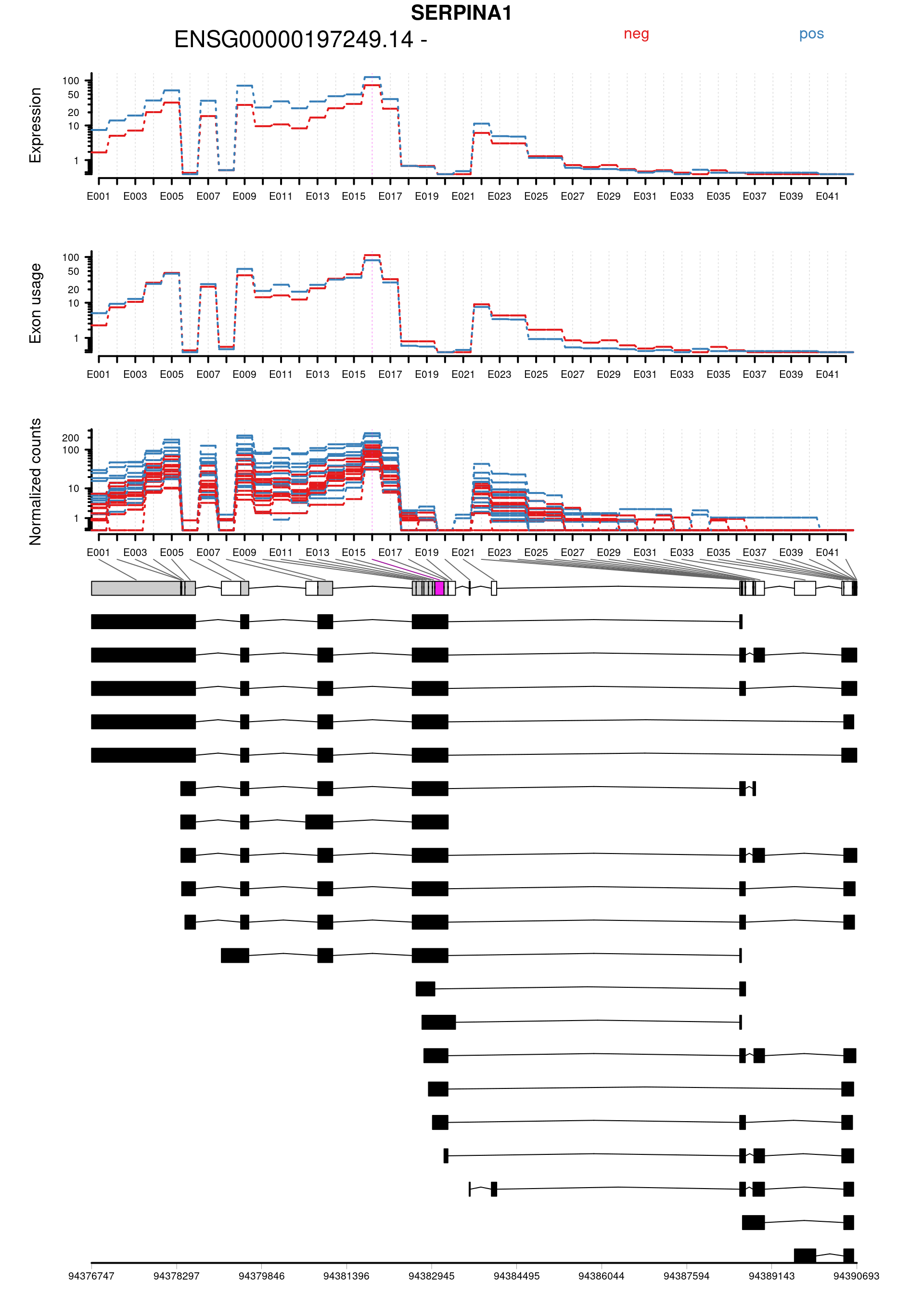
topDex[keep,] %>%
arrange(pvalue) %>%
knitr::kable()| symbol | deg | groupID | featureID | exonBaseMean | dispersion | stat | pvalue | padj | neg | pos | log2fold_pos_neg | genomicData.seqnames | genomicData.start | genomicData.end | genomicData.width | genomicData.strand | countData.CMV30 | countData.CMV31 | countData.CMV8 | countData.CMV9 | countData.CMV26 | countData.CMV14 | countData.CMV15 | countData.CMV20 | countData.CMV21 | countData.CMV1 | countData.CMV2 | countData.CMV3 | countData.CMV4 | countData.CMV10 | countData.CMV11 | countData.CMV19 | countData.CMV35 | countData.CMV51 | countData.CMV52 | countData.CMV53 | countData.CMV54 | countData.CMV56 | countData.CMV57 | countData.CMV58 | countData.CMV60 | countData.CMV61 | transcripts | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ENSG00000197249.14:E016 | SERPINA1 | TRUE | ENSG00000197249.14 | E016 | 102.353104 | 0.0059674 | 23.67040 | 1.1e-06 | 0.0323076 | 2.0440776 | 1.9363302 | -0.3616374 | chr14 | 94383003 | 94383170 | 168 | - | 120 | 93 | 38 | 162 | 112 | 116 | 55 | 68 | 99 | 60 | 86 | 51 | 189 | 111 | 93 | 243 | 97 | 131 | 24 | 175 | 128 | 74 | 164 | 110 | 69 | 19 | ENST0000…. |
| ENSG00000171793.16:E030 | CTPS1 | FALSE | ENSG00000171793.16 | E030 | 2.487082 | 0.0039669 | 23.14166 | 1.5e-06 | 0.0386619 | 0.7006737 | 0.3499062 | -1.6987830 | chr1 | 40997394 | 40997526 | 133 | + | 2 | 5 | 5 | 2 | 1 | 3 | 5 | 1 | 4 | 2 | 6 | 3 | 0 | 7 | 0 | 1 | 8 | 7 | 0 | 0 | 4 | 3 | 2 | 1 | 1 | 1 | ENST0000…. |
| ENSG00000163913.12:E012 | IFT122 | FALSE | ENSG00000163913.12 | E012 | 3.512354 | 0.0027761 | 21.39059 | 3.7e-06 | 0.0662329 | 0.7845879 | 0.4688625 | -1.3888995 | chr3 | 129451973 | 129451998 | 26 | + | 2 | 7 | 7 | 1 | 3 | 7 | 6 | 0 | 5 | 3 | 7 | 6 | 3 | 8 | 0 | 0 | 5 | 9 | 1 | 0 | 1 | 9 | 4 | 2 | 3 | 3 | ENST0000…. |
| ENSG00000136872.20:E020 | ALDOB | TRUE | ENSG00000136872.20 | E020 | 32.767507 | 0.0004712 | 21.38854 | 3.8e-06 | 0.0662329 | 1.5665267 | 1.4119173 | -0.5309091 | chr9 | 101430776 | 101430897 | 122 | - | 27 | 39 | 31 | 41 | 23 | 18 | 15 | 32 | 24 | 17 | 22 | 25 | 63 | 57 | 27 | 142 | 29 | 24 | 8 | 52 | 23 | 16 | 57 | 21 | 16 | 5 | ENST0000…. |
| ENSG00000107959.16:E024 | PITRM1 | FALSE | ENSG00000107959.16 | E024 | 5.770499 | 0.0019295 | 20.50877 | 5.9e-06 | 0.0986710 | 0.9339937 | 0.6782048 | -1.0108556 | chr10 | 3148171 | 3148291 | 121 | - | 4 | 12 | 8 | 10 | 4 | 13 | 5 | 5 | 6 | 3 | 7 | 7 | 5 | 9 | 2 | 3 | 13 | 11 | 4 | 1 | 6 | 4 | 1 | 4 | 10 | 3 | ENST0000…. |
sessionInfo()R version 4.0.2 (2020-06-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS: /config/binaries/R/4.0.2/lib64/R/lib/libRblas.so
LAPACK: /config/binaries/R/4.0.2/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] org.Hs.eg.db_3.12.0 limma_3.46.0
[3] DEXSeq_1.36.0 RColorBrewer_1.1-2
[5] AnnotationDbi_1.52.0 DESeq2_1.30.1
[7] SummarizedExperiment_1.20.0 GenomicRanges_1.42.0
[9] GenomeInfoDb_1.26.7 IRanges_2.24.1
[11] S4Vectors_0.28.1 MatrixGenerics_1.2.1
[13] matrixStats_0.59.0 BiocParallel_1.24.1
[15] patchwork_1.1.1 NMF_0.23.0
[17] Biobase_2.50.0 BiocGenerics_0.36.1
[19] cluster_2.1.0 rngtools_1.5
[21] pkgmaker_0.32.2 registry_0.5-1
[23] forcats_0.5.1 stringr_1.4.0
[25] dplyr_1.0.4 purrr_0.3.4
[27] readr_1.4.0 tidyr_1.1.2
[29] tibble_3.1.2 ggplot2_3.3.5
[31] tidyverse_1.3.0 here_1.0.1
[33] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] readxl_1.3.1 backports_1.2.1 BiocFileCache_1.14.0
[4] plyr_1.8.6 splines_4.0.2 gridBase_0.4-7
[7] digest_0.6.27 foreach_1.5.1 htmltools_0.5.1.1
[10] fansi_0.5.0 magrittr_2.0.1 memoise_2.0.0.9000
[13] doParallel_1.0.16 Biostrings_2.58.0 annotate_1.68.0
[16] modelr_0.1.8 askpass_1.1 prettyunits_1.1.1
[19] colorspace_2.0-2 blob_1.2.1 rvest_0.3.6
[22] rappdirs_0.3.3 haven_2.3.1 xfun_0.23
[25] crayon_1.4.1 RCurl_1.98-1.3 jsonlite_1.7.2
[28] genefilter_1.72.1 survival_3.2-7 iterators_1.0.13
[31] glue_1.4.2 gtable_0.3.0 zlibbioc_1.36.0
[34] XVector_0.30.0 DelayedArray_0.16.3 scales_1.1.1
[37] DBI_1.1.1 Rcpp_1.0.6 xtable_1.8-4
[40] progress_1.2.2 bit_4.0.4 httr_1.4.2
[43] ellipsis_0.3.2 pkgconfig_2.0.3 XML_3.99-0.5
[46] dbplyr_2.1.0 locfit_1.5-9.4 utf8_1.2.1
[49] tidyselect_1.1.0 rlang_0.4.11 reshape2_1.4.4
[52] later_1.1.0.1 munsell_0.5.0 cellranger_1.1.0
[55] tools_4.0.2 cachem_1.0.4 cli_3.0.0
[58] generics_0.1.0 RSQLite_2.2.5 broom_0.7.4
[61] evaluate_0.14 fastmap_1.1.0 yaml_2.2.1
[64] knitr_1.31 bit64_4.0.5 fs_1.5.0
[67] whisker_0.4 xml2_1.3.2 biomaRt_2.46.3
[70] compiler_4.0.2 rstudioapi_0.13 curl_4.3
[73] reprex_1.0.0 statmod_1.4.35 geneplotter_1.68.0
[76] stringi_1.5.3 highr_0.8 lattice_0.20-41
[79] Matrix_1.3-2 vctrs_0.3.8 pillar_1.6.1
[82] lifecycle_1.0.0 bitops_1.0-7 httpuv_1.5.5
[85] R6_2.5.0 hwriter_1.3.2 promises_1.2.0.1
[88] codetools_0.2-18 assertthat_0.2.1 openssl_1.4.3
[91] rprojroot_2.0.2 withr_2.4.2 Rsamtools_2.6.0
[94] GenomeInfoDbData_1.2.4 hms_1.0.0 grid_4.0.2
[97] rmarkdown_2.6 git2r_0.28.0 lubridate_1.7.9.2